Ph.D. Completed in 2022. My research is involved with theory and computer simulation on the reactions, energy transfer, and dynamics of polyatomic molecules. This includes the construction of analytical global potential energy surfaces (PES) for polyatomic systems based on the double many-body expansion (DMBE) developed by Varandas and collaborators. In addition, high-level ab initio electronic
Ph.D. Completed in 2022. My research is involved with theory and computer simulation on the reactions, energy transfer, and dynamics of polyatomic molecules. This includes the construction of analytical global potential energy surfaces (PES) for polyatomic systems based on the double many-body expansion (DMBE) developed by Varandas and collaborators. In addition, high-level ab initio electronic
structure calculations such as MRCI+Q, CASPT2, and CCSD(T) are performed using the MOLPRO program. Once a reasonable PES is constructed it can be used to describe several physical and chemical processes such as plasma physics, spectroscopic constants, reactions, and rovibrational energy transfer processes. For dynamics, the framework of the quasi-classical trajectories (QCT) methodology is used. Such procedure has been pretty used since the first works from Karplus et al. being these fundamentals for the development of molecular dynamics. Alternatively to an analytical representation of potential energy, an artificial neural network (NN) approach has been proposed for the PE curves in diatomic systems using open source library
FANN (Fast Artificial Neural Network). Currently, I am working in the investigation of translation-to-vibration energy transfer (T-V ET) process in hydrogen (or argon) atom and sulfur dioxide molecule.
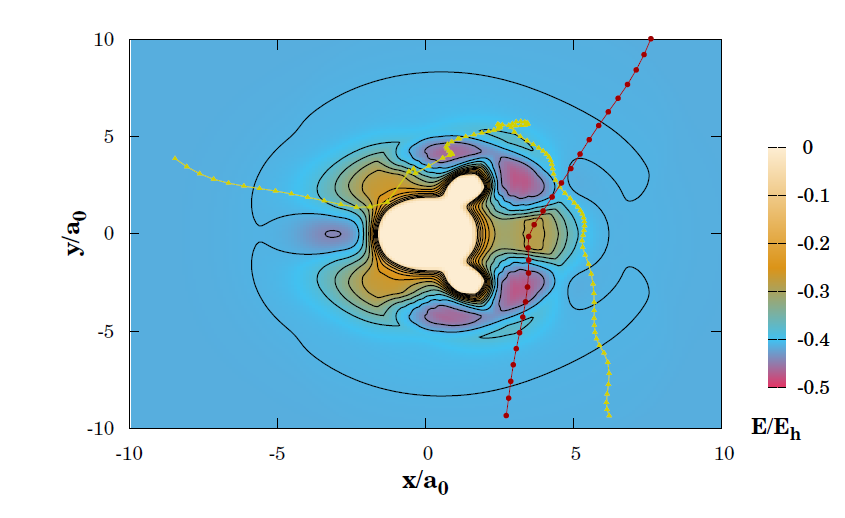
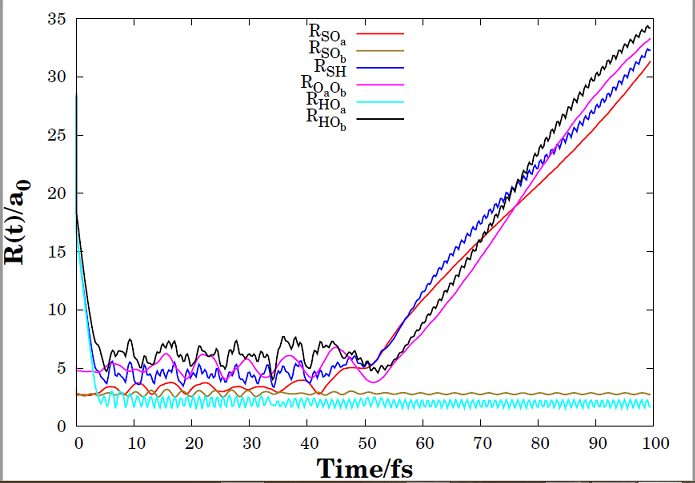
For H+SO2 reaction, a six-dimensional double many-body expansion (DMBE) potential energy surface (PES) for the ground electronic state of HSO 2 was used to represent the interatomic interactions. It should be noted that this PES was previously reported by Ballester and Varandas. As results, the calculations reproduce the experimental evidence that during majority of inelastic non-reactive collision processes, there is a metastable intermediate formation (HOSO or HSO2 ). Nevertheless, the analysis of the trajectories shows that there are two distinct mechanisms in the T-V ET process: direct and indirect. Direct T-V processes are responsible for the high population of SO2 with relatively low vibrational excitation energy, while indirect ones dominate the conversion from translational energy to high values of the vibrational counterpart.
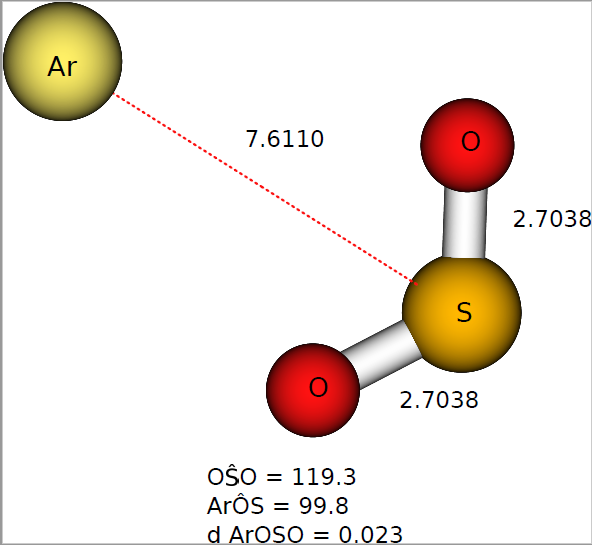
In Ar + SO 2 reaction, A new realistic potential energy surface (PES) for the ArSO 2 system was developed using a pairwise addition for the four-body energy term within the double many-body expansion. From dynamical calculations, the cross sections for the T-V excitation process indicate a barrier-type mechanism due to strong repulsive interactions between SO 2 molecules and the Ar atom. Corrections to zero-point energy (ZPE) leakage in QCT were carried out using VEQMT c and NVEQMT methods. Rate coefficients and cross sections are calculated for some vibrational transitions using pseudo quantization approaches (Histogram and Gaussian Binning) of the vibrational energy of products.